A.4.
Specific Aim 2: Integration of the Approaches Developed for Different Scales
A.4.1.
Breadth of molecular and mathematical approaches.
The development of physically realistic but mathematically tractable models has
been a major goal in the uppermost (mathematical) and lowermost (molecular)
approaches, in particular. Extensive progresses have been made in these two
groups of studies in the last three decades (see Table A.2). We can refer to two reviews from the
pre-NPEBC leading PIs at these two levels (103;106), which
provide an overview of the models (discrete and continuous) and methods
(analytical and numerical) that have been developed by molecular (106) and
mathematical (103) modelers
for studying macromolecular and cellular dynamics, respectively. A recent paper
by Bahar and coworkers also typifies the efforts (of molecular modelers) for
bridging between low resolution analytical approaches and full atomic MD simulations,
capturing commonalities, assessing what is being neglected at the higher scale (114). Given
the extensive work already accomplished by molecular and mathematical modelers,
we will focus here on the approaches for filling the intermediate gap, mainly bridging
(i) between mathematical models of cellular networks and stochastic (MC)
simulations of microphysiological events, on the one hand, and (ii) between the
latter MC simulations and the molecular simulations and analytical methods, on
the other. In a broad sense, a two-level approach is adopted, in which the
complex signaling networks are modeled in terms of effective control motifs or
“circuit” modules, and these modules are modeled using the molecular
biochemical and biophysical properties of their elements, as suggested by
Asthagiri et al. (115).
A.4.2. Modular Control Theory Analysis of Biochemical
Networks. Even relatively small metabolic pathways such
as glycolysis can be shown to exhibit chaotic behavior in some regions of
parameter space (116). Dynamical systems analysis of
simplified network modules (e.g., positive and negative feedback loops) has
shown that important properties like adaptation, oscillation, and bistability
can emerge from such simple cross-interactions of a small number of signaling
molecules (117-119).
Bifurcation analysis has been used with remarkable success to show how
interactions between cyclin-dependent kinases and their associated proteins
control the timing of eukaryotic cell division during early embryogenesis (120).
Such studies have shown how positive and negative feedback loops
involving Cdc25, Wee1, APC (anaphase-promoting complex) and MPF
(M-phase-promoting factor) regulate MPF activation and degradation (121;122), and in more general terms, how
dynamic systems theory applied to small modules or subnetworks (or control
motifs) that are amenable to theoretical analysis (119), can establish the connection
between molecular regulatory mechanisms and cell physiology (115;118;123). These modules are ideally suited
for simulations with the adapted forms of MCell (see below). We will explore
the dynamics of such modules, or control motifs, both by simulations and
analytical methods, and combine these into the large scale network.
The
synchronization of the subsystems or the timing
of the signals they transmit can, however, have synergistic effects far more
complex than those expected from simple additive rules. For example networks
have properties –extended signal duration, feedback loops, stimulation
thresholds, or multiple signal outputs - that individual pathways do not have (124). Likewise
groups of coupled control motifs in complex networks can exhibit distinctive
features. We anticipate several cycles of refinement between simulations and
customized assays so as to build integrated models that include such coupling
effects.
A.4.3.
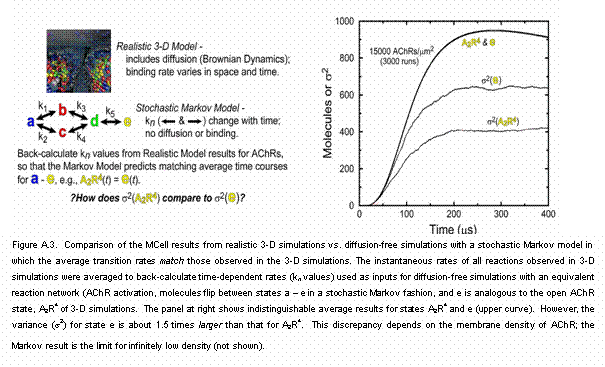
Counter-intuitive Effects of Realistic Spatial Organization (Figure A.3). Two counter-intuitive
examples taken from our MC simulations of miniature endplate currents (mEPCs)
illustrate the importance of considering space-dependence. We simulated the
reversible activation of muscle acetylcholine receptors (AChRs) by the binding
of two molecules of diffusing ACh, with subsequent conformational change from
the closed state (A2R3) to an open-channel state (A2R4)
that produces the mEPC. The observations are: (i) Adding a JF to an otherwise
flat synaptic cleft effectively doubles the concentration of AChRs close to the
mouth of the fold, and increases the local cleft volume by a factor of
~1.5. Intuition predicts an increase in mEPC amplitude. However, simulations show a decrease, due to non-linear dynamic
effects in the mEPC rising phase. (ii) One might expect that spatially
realistic MC simulations would lead to greater noise (variance, s2) in the time course of the reactant amount in a given state,
compared to matching simulations in which there is no explicit space. However, Fig. A.3 shows the opposite can be true in a realistic
subcellular environment, and that the discrepancy can be substantial. Such
counter-intuitive results may have dramatic implications for qualitative and
quantitative predictions of steady state, adaptive, oscillatory, and chaotic
behaviors of biochemical networks.
A.4.4. Integration of Cellular and Microphysiological Models. The aim of these
studies will be to characterize the effects of stochastic and spatial
parameters on the dynamics of abstract
network motifs, and to develop heuristic rules and functions that can be
added to cellular models based on differential equations. By abstract
network motifs we mean generic positive and negative feedback loops (and
combinations thereof) that appear in virtually all biochemical networks of
interest, including the three developmental projects DP1-3. Our general method
will involve the formulation of several models for the same biological
questions, defined at different resolutions, for studying the limitation at
each level as well as their relations. For example, consider four levels of
modeling: discrete particles with space dependence (e.g. MC simulations);
discrete particles/probabilities, no space dependence (e.g. master equation
formalism); continuum models with PDEs (a limit in the first model), and
continuum with ODEs (a limit of the second model). A specific study may be:
1)
Identify or postulate
a control motif in the network of interest, and build its block diagram based
on experimental information.
2)
Analyze the
predictions of (1) using continuum models and phase plane and/or bifurcation
analysis as allowed by the problem's dimensionality.
1)
Examine the dynamics
of the same motif with discrete modeling using MCell and "identical"
initial conditions: (a) well-mixed reactants (uniform concentration) in a
symmetrical unit volume (Fig. A.4), (b) well-mixed reactants in different
volumes, i.e. varying the number of
reactants, (c) varying the mobility of the species, while  maintaining average binding rates (16), (d) holding the total number of reactants fixed, but
varying their spatial distributions, and (e) repeating the simulations with the
above mentioned stochastic Markov model.
maintaining average binding rates (16), (d) holding the total number of reactants fixed, but
varying their spatial distributions, and (e) repeating the simulations with the
above mentioned stochastic Markov model.
2)
Study the passage from
discrete or hybrid models to continuous models inasmuch as possible, and
develop heuristic rules that relate spatial parameters, reactant numbers, and
reactant mobilities to network behavior, so that the mathematical models can be
adapted to make similar qualitative and quantitative predictions. Hybrid modeling
methods that combine the classical approach of differential equations with
stochastic and MC methods (125) will also be considered for this
aim.
3)
Tune model parameters with experimental
data using inverse problem formulation via optimization techniques. This allows
efficient sampling of the parameter space and saves the need to reiterate
similar models many times. Such approaches have been done successfully in
material science, aerodynamics shape design and more. Our techniques for
optimization and inverse problems (126-128) will be used for that part.
4)
If agreement with experimental data
is not satisfactory after optimal values have been selected in step 5,
reevaluate the model system assumptions, and reiterate steps 1-5.
MCell
is presently capable of simulating arbitrary reactions between diffusing small
molecules and static binding sites (receptors) on reflective, transparent, or
absorptive surfaces. Step 3 above can be
begun with present capabilities, but will also require simulation of interactions
between diffusing molecules themselves. This extension of MCell is presently
being pursued. Step 3 will also ultimately lead to large-scale parameter
sweeps, and the resources and support of PSC will be a major factor that will
ensure high throughput. In addition,
MCell use is now being extended to Grid-based computation, with NSF support
(ITR0086092). This is a collaborative
project between MCell developers, M. Ellisman (Neuroscience, UCSD), and
grid-based distributed computation experts (F. Berman (Project Director), H.
Casanova, R. Wolski, UCSD; J. Dongarra, Univ. Tennessee Knoxville). The Computational Grid is an emerging
platform for deployment of large-scale scientific and engineering projects. MCell is the chosen prototype application for
this project, because large sets of simulations can generally be run in
parallel with different ranges of spatial and kinetic parameter sweeps and
sensitivity analyses.
Developing large scale models from a
microscopic one is a hard question in general. The applications we are
considering here may benefit from experience obtained over many years in other
fields. A classical example is the emergence of the Navier-Stokes (NS)
equations from molecular motion modeled by Newton’s equations of motion. An
interesting lesson from this example is that macroscopic variables may have different
forms than their
microscopic counterparts (e.g. temperature
in NS equations, accounted for by velocity distributions on the particle
level). Thus, the set of proper variables to use on each level is a most
difficult question. Additional lessons can be learned from the coarse-graining
processes that lead to macroscopic equations. The passage to larger scales uses
probability distributions and their dynamical equations, the Boltzmann equation
being the most obvious example. This supports a description of the system in
terms of probability functions, in addition to the other approaches mentioned.
Averaging techniques applied to such models result in continuum models.
A.4.5. Integration of Microphysiological and Molecular
Models. As mentioned above, there have been numerous
studies by the pre-NPEBC investigators, aiming at developing models and tools
that can explore the mesoscale dynamics. Two noteworthy examples are, the
recent extension of the elastic network models to multimeric complexes of 104
-105 residues (91;92), and the solution of the
Poisson-Nernst-Planck PDEs to compute the current-voltage characteristics of
ion channels (30). The former is a purely mechanical approach for a collection of discrete interaction sites, while the
latter resorts to a continuum
description of permeating ions in a self-consistent mean-field of electrostatic interactions. These are
therefore quite different, in a sense orthogonal, approaches, and both have
approximations, adopted for mathematical simplicity or computational
efficiency. The former neglects chemical specificities or energetics, while the
latter cannot address the effects of finite size or excluded volume in the case
of narrow channels. Yet, both approaches yield results in quantitative agreement
with experimental data, and provide insights as to the dominant mechanisms of
collective dynamics. And to address the limitations of these approaches, a
strategy is to perform simulations. BD simulations of the same ion channel were
indeed performed to this aim (129), and likewise, the GNM results were
compared with those from MD simulations coupled with essential dynamics
analysis (115).
This type of cross-examination of a given system by both more detailed
and reduced methods has proven useful for assessing the most important
‘details’ that need to be incorporated in the higher level models and
methods. An even more synergistic
interaction between the two orthogonal approaches would to combine in a hybrid model the substates and
fluctuations information.
Our
goal is to extend this type of biochemical and biophysical interactions to even
higher scales, and merge with MCell-type simulations. There are many possibilities for such integrating approaches,
and a major aim of planning studies will be to identify the most needed and
feasible ones. Examples are;
1)
Cell signaling and
regulation dynamics are generally dependent on the assumed molecular substates
and rate constants for the passages between them (130). Coarse-grained molecular models coupled with analytical
tools such as mode analysis, as developed by the pre-NPEBC investigators (Table
A.2), could provide information on the dominant substates, their relative free
energies, and their fluctuation or transition rates.
2)
Coarse-grained
molecular studies could also be used to provide membrane properties or charge
distributions, which in turn could be implemented as new empirical features in
microphysiological simulations. Eventually, it should be possible to
superimpose energetic effects (empirical force fields at the cellular scale) on
the presently random displacements of molecules in MCell, based on the data
provided by theoretical and experimental studies at the molecular level.
3)
Molecular simulations
could be used to provide surface topologies and properties of macromolecular
complexes, which then would be incorporated into microphysiological simulations
as mesh objects.
4)
Coarse-grained
molecular studies could ultimately provide input to microphysiological
simulations based on volume meshes rather than surface meshes, and which thus
could combine present Monte Carlo algorithms with finite element methods. It is a challenge,
however, to select the structural and/or spatial characteristics at an optimal level of detail, given the
trade-off between accuracy and computational efficiency.
Finally,
we need to further develop the analytical tools for the interpretation and
generalization of the results. The software developed by Ermentrout (7) is an excellent resource for analyzing mathematical models.
Interestingly, models and methods used by molecular and mathematical modelers
share several common features: For example, MC methods are used both for
simulating microphysiological processes (16;17) and macromolecular
dynamics (131); (ii) the network connectivity
is an important determinant of dynamics both at the cellular level (between
interacting molecules) and at the molecular structural model (between
residues), (iii) the master equation formalism has been advantageously used for
exploring macromolecular dynamics (132;133) (106), protein folding kinetics (134), or biochemical networks (96;
97;105;135;136), and same methods of analysis based on eigenvalue
decomposition hold for all cases. These examples suggest that the mathematical
tools that we have developed for analyzing the dynamics of intramolecular
interactions, can be applied, with suitable redefinition of system parameters,
to intermolecular or cellular networks, and vice versa.