
Accumulation of glutamate at extracellular (EC) space above physiological (micromolar) levels may cause neurotoxic effects. The
concentration of glutamate in the chemical synapse may increase by 103-104 fold succeeding its release triggered by an action potential,
and it is critical to have a mechanism in place to clear or shuttle the excess glutamate. This is where the glutamate transporters (GluT)
come into play. GluTs are trimeric membrane proteins located on neurons and glia (astrocytes). Their precise functioning (uptake
and re-uptake of glutamate) is essential for regulating the cycle of signaling, or terminating the synaptic transmission. They not only
maintain the glutamate concentration below excitotoxic levels but also regulate the activity of the glutamate receptors in the synapse:
they prevent sustained activation and thereby 'desensitization' of ionotropic receptors, and influence glutamatergic transmission by
metabotropic receptors. GluTs also assists in regulating the glutamate-glutamine cycle, i.e. conversion into glutamine in the glia,
succeeded by the transport to neurons to be converted back to glutamate.
Related Publications
Mechanism of extracellular gate opening in outward facing GltpH (pdf)
Mechanism of substrate release in inwards facing GltpH (pdf)
Large Scale motions regulate functional properties of GltpH (pdf)
For movies and more information on Glutamate Transporter simulations (click here)

Glutamate Receptors
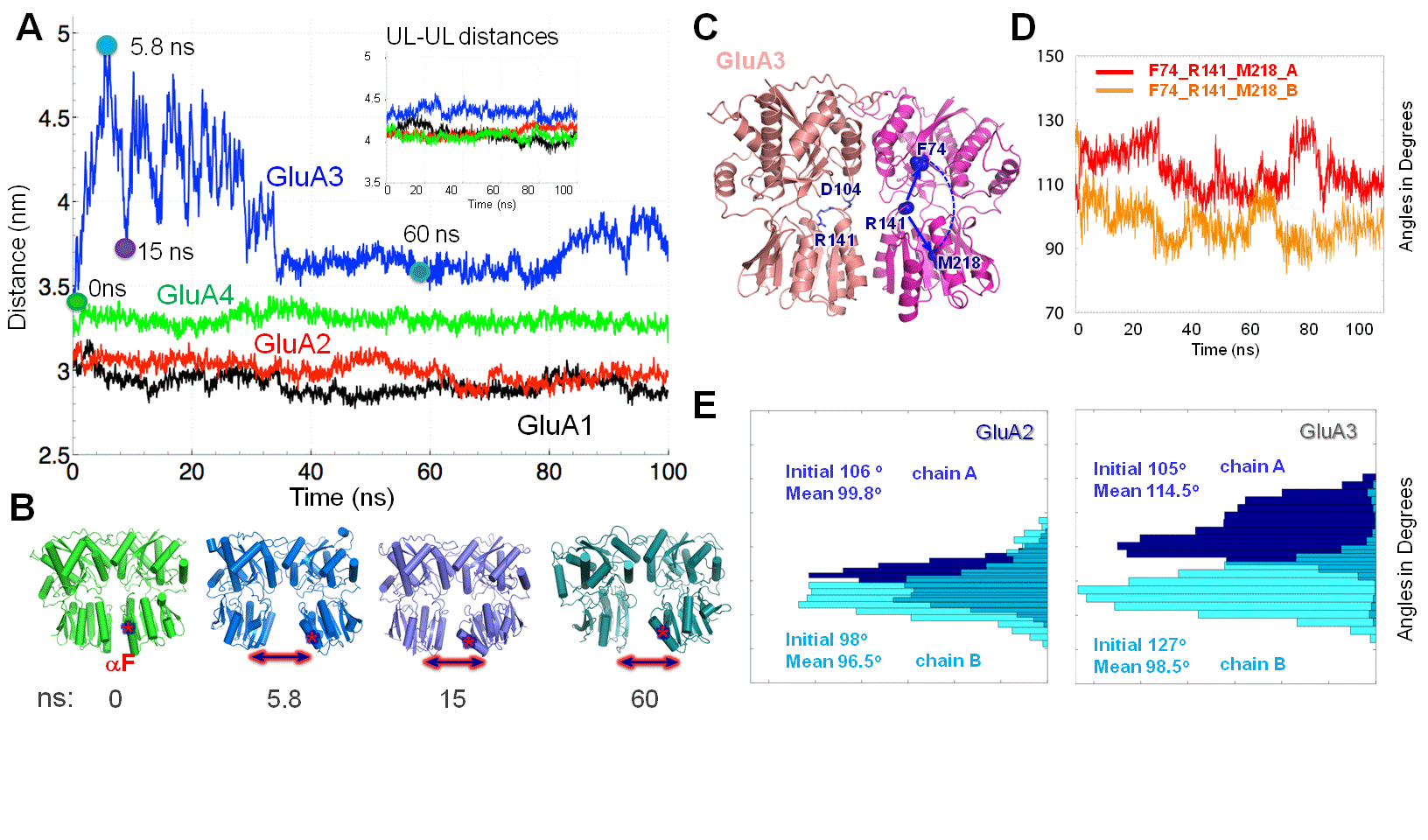
Ionotropic glutamate receptors harbor two domains in their extracellular region, the membrane-proximal ligand-binding domain (LBD)
and the distal N-terminal domain (NTD). These are involved in signal sensing: the LBD binds L-glutamate, which activates the receptor
channel. Ligand-binding to the NTD modulates channel function in NMDA receptors, which has not been observed for AMPA receptor (AMPAR) NTDs.
Structural data suggest that AMPAR NTDs are packed into tight dimers and have lost their signaling potential. We have characterized NTD dynamics
from both AMPAR- and NMDAR- subfamilies using a variety of computational tools leading to a description of the motions which underly NMDAR NTD allosteric
signaling. Unexpectedly, AMPAR NTDs are capable of undergoing a similar dynamic spectrum; although dimerization imposes restrictions the two subfamilies
sample similar,interconvertible conformational subspaces. Finally, we solve the crystal structure of the AMPAR GluA4 NTD, combined with all-atom molecular
dynamics simulations we characterize regions pivotal for an as yet unexplored dynamic spectrum of AMPAR NTDs.
Related Publications
Dynamics and allostery in N-terminal domain of iGLuR2 (pdf)
Comparison of dynamics of NMDA- and AMPA N-terminal domains (pdf)
MD movie of NTD-interface destabilization 
HIV-glycoprotein gp120
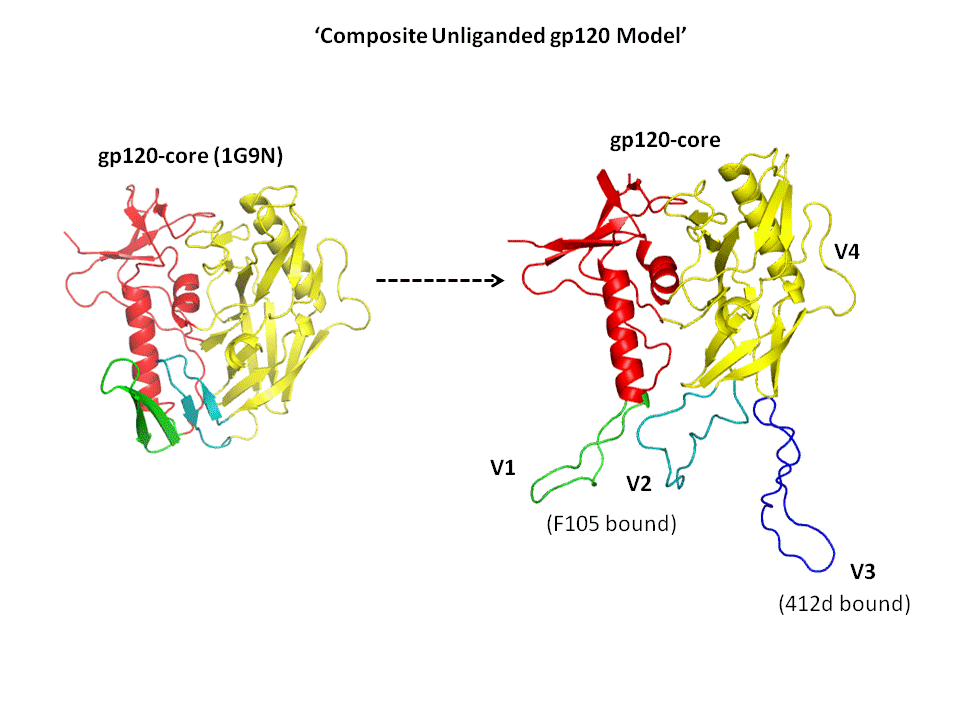
Infection of HIV-1 begins with a series of dynamic binding events between the trimeric glycoprotein envelope spike and the host cell
CD4 and chemokine receptors. The envelope trimer (gp160) is composed of three gp120 glycoproteins and the three transmembrane gp41 proteins.
The first dynamic event occurs via binding of gp120 to the host T-cell CD4 receptor, followed by extensive restructuring of gp120.
This conformational change results in the exposure of the chemokine binding site on gp120, thus permitting binding to either of the chemokine
receptors. Upon chemokine receptor binding a second conformational change occurs in the gp41 to form the fusion peptide that inserts in the host
cell membrane, leading to viral entry. The CD4 induced gp120 conformational change has been characterized thermodynamically, showing a highly
favorable binding enthalpy balanced with a highly unfavorable molecular ordering. This thermodynamic signature resembles protein folding, rather
than binding, and reflects the large molecular ordering of gp120 upon CD4 binding. A similar thermodynamic signature is exhibited by soluble
CD4 (sCD4) binding to both full-length gp120 (gp120full) and the truncated core gp120 (gp120core).
Related Publications
Communication propensities in gp120 (pdf)
Enhanced dynamics upon ligand binding in gp120 (pdf)
Spontaneous rearrangement of Beta strands 20/21 in unliganded gp120 (pdf)
MD movie of gp120 Spontaneous Rearrangement
|
| |
RTX-Toxins
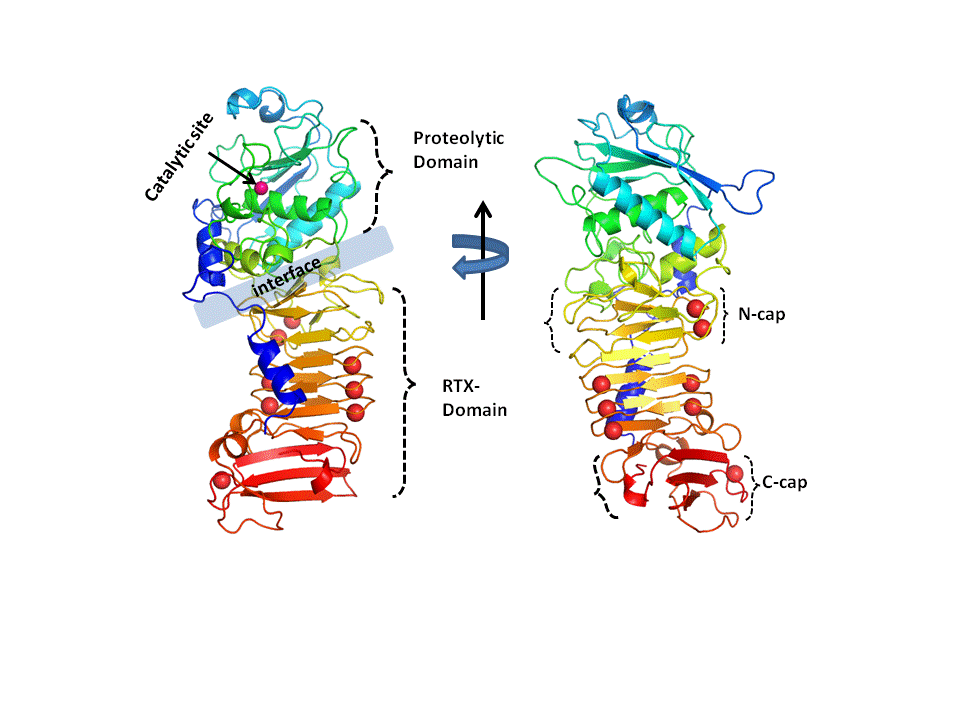
Pseudomonas aeruginosa (PA) is an opportunistic pathogen that contributes to the mortality of patients with cystic fibrosis (CF), burn victims
and immune-compromised individuals. The virulence of PA is mediated by the secretion of virulence factors such as repeat-of-toxin(RTX)
proteins. Alkaline protease (AprA), one such RTX-protein secreted by PA is implicated in multiple modes of bacterial infection. The protease
is secreted in an unfolded form into the extracellular space, where it undergoes transition to a functional, folded form. The disorder-to-order
transition of the RTX-domain has been shown to be mediated by Ca2+ binding and the folded RTX-domain itself is proposed to facilitate proteolytic
domain (PD) folding, via the PD/RTX-domain interface. The underlying detailed mechanism of the toxic activities of RTX-toxins remains to be
elucidated. What is known however is that the intrinsic disorder of the RTX-domain in the bacterial cytosol facilitates secretion and that the
unfolded form of the RTX-toxin limits the enzymatic activity. The identity of structural elements of the protease which are required for regulation
of protein folding is a topic of investigation. These studies are aimed at identification of specific structural elements that stabilizing the protein
fold. This information can be used to design exepriments to characterize structure and function of misfolded proteins.
|
| |
HSV-1 glycoprotein gH1
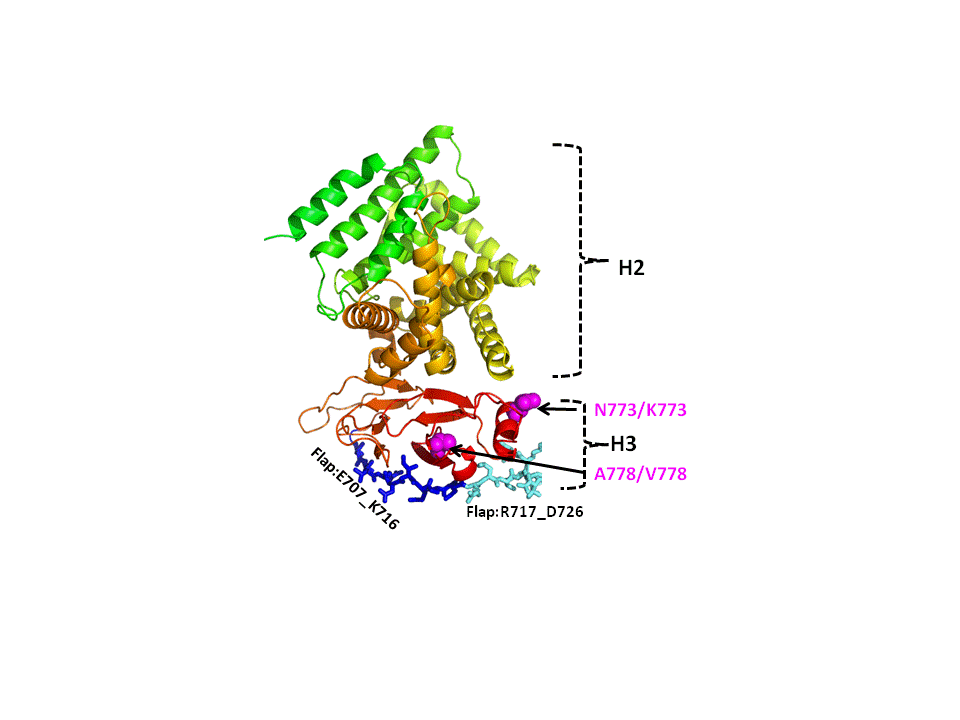
Over the last decade and a half, herpes simplex virus type 1 (HSV-1) has been developed into a leading gene therapy vector system.
Its large double-stranded DNA genome can simultaneously accommodate multiple therapeutic genes and the virus has broad cell and tissue
tropism, enabling its application to many different diseases. Recent advances have produced HSV-1 vectors that are limited in their infectivity
to one or a few cell types, advancing the prospect that invasive vector injection at the disease site can be replaced with systemic application.
Our goal is to produce viruses that are optimally infectious for their specific target cells or organs to increase their therapeutic efficacy
on systemic delivery. To this end we have isolated gain-of-function mutations in several of the HSV-1 glycoproteins that act in concert to
mediate virus entry into cells, gD, gB, gH and gL. We seek to understand the structural basis of the entry-enhancing effects
of mutations in gH using the published structures of gH from related herpesviruses, particularly HSV-2, for guidance. An understanding of the
structural consequences of these mutations will facilitate the rational engineering of gH and its direct partners in entry, gD and gB, to
accomplish further gains in entry efficiency. These studies promise to provide novel information about the key interactions between
the HSV-1 entry glycoproteins, which may lead to the design of new and highly specific anti-viral drugs.
| | | | | 
